Gaschromatographen für Ausbildung und Industrie
Grundlagen der Gaschromatographie
Die Chromatographie in der Gasphase ist eine der Säulen der instrumentellen Analytik. Nach einer stürmischen technischen Entwicklung in den 70er und 80er Jahren hat sich diese Methode auf einem hohen Niveau etabliert. Für alle Stoffgemische geeignet, die sich ohne chemische Veränderungen verdampfen lassen, gestattet sie die Trennung selbst komplizierter Substanzgemische in sehr kleinen Stoffportionen. In vielen Fällen kann über die Retentionszeiten eine Identifizierung einzelner Komponenten erfolgen; aus den Peakflächen ist nach Eichung eine quantitative Bestimmung möglich.
Das Funktionsprinzip
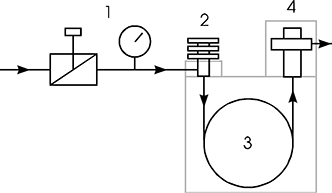
Ein inerter Trägergasstrom, meist Wasserstoff oder Helium, durchströmt nacheinander eine Druckregeleinrichtung (1), den Injektor (2) die Trennsäule (3) sowie den Detektor (4). Das zu trennende Stoffgemisch wird mit einer Mikroliterspritze in den beheizten Injektor eingebracht und dort verdampft. Die gasförmigen Komponenten der Mischung werden vom Trägergas in die Säule und später in den Detektor transportiert. In der Säule selbst findet die räumliche Trennung der gasförmigen Komponenten statt, indem diese in der Säule unterschiedlich lange festgehalten werden. Durch diese Retention, (lat: retinere = zurückhalten) treten die einzelnen Komponenten zu unterschiedlichen Zeiten aus der Säule aus. Der Detektor liefert nun ein elektrisches Signal, wenn eine Komponente der zu trennenden Mischung aus der Säule austritt. Das Detektorsignal wird in Abhängigkeit von der Zeit aufgezeichnet, früher mit einem Laborschreiber auf Papier, heute wird das Signal digitalisiert und auf gängigen Speichermedien zusammen mit den übrigen Messdaten archiviert.
Das chromatographische Trennprinzip
Verteilung
Das Phänomen der Verteilung lässt sich am besten durch einen kleinen Versuch verdeutlichen: In eine Kaliumjodidlösung der Konzentration c=1mol/l werden einige Kristalle Jod gegeben.
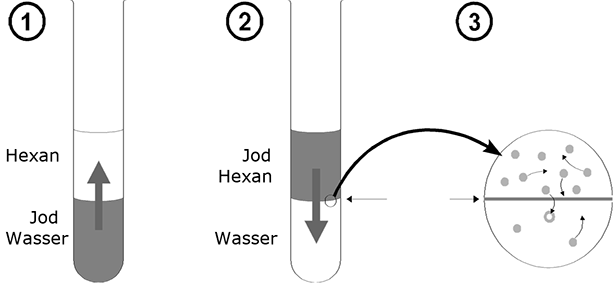
Unter Schütteln löst sich das Jod mit brauner Farbe. Überschichtet man diese Lösung mit Hexan, so bemerkt man, dass die Jodteilchen durch die Phasengrenze in das Hexan diffundieren und diesem eine violette Farbe verleihen. Nun wird der Versuch umgekehrt: Man löst etwas Jod in Hexan und unterschichtet mit KJ-Lösung. Jetzt diffundiert das Jod von der Alkyl-Phase in die wässrige Phase. Diese beiden Ausgangspositionen führen in klassischer Weise zu einem dynamischen Gleichgewicht. Wartet man in beiden Fällen genügend lange, so ist die Zahl der Phasendurchtritte von "oben" nach "unten" gleich pro Zeiteinheit, d.h. an den Konzentrationen der Teilchen in den beiden Phasen ändert sich nichts mehr.
Nach NERNST gilt in diesem Fall für kleine c:
c(Jod in Phase 1) / c(Jod in Phase 2) = const
Das heißt: Das Verhältnis der Konzentrationen ist konstant. Diese Konstante wird auch Verteilungskoeffizient genannt.
Verteilung bei bewegten Phasen
In einem Gedankenexperiment reihen wir nun eine größere Anzahl Reagenzgläser der obigen Art aneinander, nehmen aber zusätzlich an, dass eine der Phasen seitlich zu verschieben sei (mobile Phase), die andere aber fest ist (stationäre Phase). Desweiteren soll die Bewegung der mobilen Phase langsam im Vergleich zur Geschwindigkeit der Diffusionsvorgänge sein.
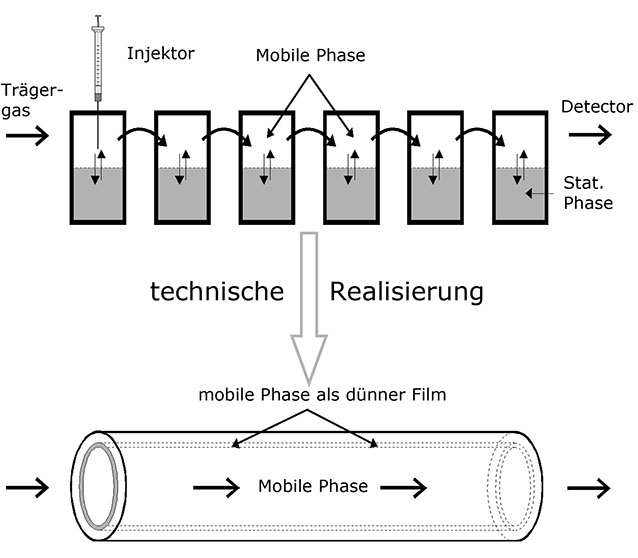
Dieses Gedankenexperiment liefert mehrere Ergebnisse:
• Die Teilchen in einem solchen System wandern mit der mobilen Phase, jedoch langsamer als diese, da sie nur dann bewegt werden, wenn sie gerade in der mobilen Phase sind.
• Der Bereich, in dem die zu trennenden Teilchen durch die Säule wandern, wird mit der Dauer des Experiments breiter.
• Die Geschwindigkeit der Teilchen relativ zur mobilen Phase hängt direkt vom Vertei-lungskoeffizienten ab, denn ein Teilchen, das sich statistisch gesehen öfter in der stationären Phase aufhält, ruht öfters. Daher ist es im Durchschnitt langsamer als ein Teilchen, welches häufiger in der bewegten Phase zu finden ist. Die Relativge-schwindigkeit der Teilchen ist also stoffabhängig und kann daher zur analytischen Trennung benutzt werden.
Der Verteilungskoeffizient hängt sehr stark von der Temperatur ab. Dieser Mechanismus ist leicht einzusehen, denn mit zunehmender Temperatur nimmt die Teilchenbewegung zu und damit die Tendenz, sich im gasförmigen Zustand (dem Zustand größerer Entropie) aufzu-halten. Diese Tatsache wird in der Chromatographie ausgiebig benutzt.
Hat man in einer zu trennenden Mischung Komponenten mit stark unterschiedlichen Siede-punkten, so kann bei konstanter Säulentemperatur unter Umständen keine Trennung in alle Einzelstoffe erzielt werden. Entweder werden bei niedriger Säulentemperatur die höhersiedenden Stoffe erst nach sehr langer Zeit aus der Säule austreten (eluiert werden) oder aber es werden bei hoher Säulentemperatur die niedrigsiedenden Komponenten nicht getrennt. Abhilfe schafft hier eine Temperaturprogrammierung des Säulenofens.
Meist wird in der Praxis die Messung mit einer Säulentemperatur gestartet, die auf die am leichtesten flüchtige Komponente abgestimmt ist. Nach einer Phase konstanter Temperatur (t1-t0) wird nun mit einer linear mit der Zeit zunehmenden Säulentemperatur gearbeitet, (t2-t1) so dass auch die höchstsiedenden Komponenten aus der Säule eluiert werden. Nach der linearen Aufheizung wird meist noch mit einer konstanten Temperatur weiter gearbeitet. (t2-Ende) Eine solche Temperatur Programmierung des Säulenofens ist heute allgemein Stand der Technik.
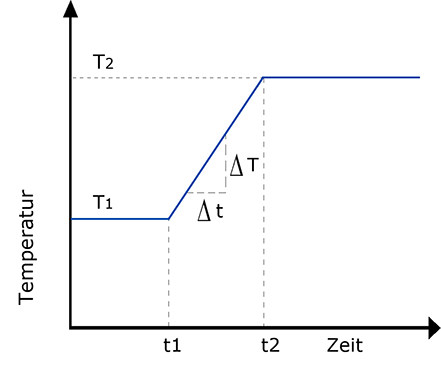
Ein typisches Temperaturprogramm zeigt Abb. 21. Die Aufheizgeschwindigkeit ∆T/∆t wird in °C/min oder K/min angegeben; übliche Werte liegen zwischen 1 und 10 K/min.
Home
Kontakt
Impressum / Datenschutzerklärung