Gaschromatographen für Ausbildung und Industrie
1. Gepackte Säulen
Bei diesem Säulentyp enthält eine Röhre aus Chromstahl oder Glas ein poröses Material mit einem Partikeldurchmesser von 0,05 bis 0,5mm als Trägersubstanz, die mit der stationären flüssigen Phase beschichtet ist. Die große Oberfläche dieses Stoffes erlaubt die schnelle Einstellung des Diffusionsgleichgewichts zwischen der mobilen Gasphase und der stationären flüssigen Phase. Der Massenanteil an flüssiger Phase variiert abhängig von der Anwendung zwischen 0,5 und 25%. Gepackte Säulen enthalten wesentlich mehr flüssige Phase als Kapillarsäulen und sind daher gut geeignet zur Trennung leicht flüchtiger Komponenten. Auch ist die Trennbarkeit größerer Probenmengen von Vorteil, insbesondere in Verbindung mit einem Wärmeleitfähigkeitsdetektor (WLD), dessen Empfindlichkeit prinzipbedingt kleiner ist als diejenige anderer Detektoren wie zum Beispiel des Flammenionisationsdetektors (FID). Das vorliegende Gerät enthält in der Standardversion 1 eine gepackte Säule aus Chromstahl mit einem inneren Durchmesser von 2 mm und einer Länge von 2 m. Sie enthält als Träger-material ein Präparat aus Diatomeenerde mit einer Korngröße von 80-100 mesh und einer Beschichtung von 5 % Polydimethylsiloxan (OV-1).

schematischer Aufbau einer gepackten Säule
2. Kapillarsäulen
Diese Säulen bestehen heute meist aus einem dünnwandigen Rohr aus reinem Quarz SiO2, im englischen mit Fused Silica (FS) bezeichnet. Ebenfalls aus dem englischen Sprachraum stammt die Abkürzung OTC (open tubular column) für diesen Säulentyp. Die stationäre Phase ist hier in Form eines dünnen Films auf die Innenwand der Röhre aufgebracht. Oft wird dieser Film auch durch chemische Bindungen an das Siliciumdioxid fixiert. Dadurch lassen sich die Säulen zur Reinigung mit geeigneten Lösungsmitteln spülen, ohne dass die Belegung herausgelöst wird. Zum mechanischen Schutz sind die Säulen meist mit einem Überzug aus dem Kunststoff Polyimid versehen. Kommerziell hergestellte Kapillarsäulen sind in abgestuften Abmessungen des Innen- bzw. Außendurchmessers erhältlich:
Innendurchmesser Aussendurchmesser
0,10 mm 0,27 mm
0,20 mm 0,35 mm
0,25 mm 0,38 mm
0,32 mm 0,48 mm
0,53 mm 0,75 mm
Die Filmdicken der stationären Phase variieren zwischen 0,1 und 5 Mikrometer. Gängige Säulenlängen liegen zwischen 10 und 50 Metern. Die obengenannten Gesetzmäßigkeiten bezüglich der Verteilung gelten nur näherungsweise für kleine Konzentrationen. Überlastet man eine Säule, indem man viel Substanz zu trennen versucht, so erhält man verzerrte Peaks und damit eine Einbuße an Trennleistung. Es ist leicht einzusehen, dass Säulen mit dicker stationärer Phase mehr Analysensubstanz vertragen. Andererseits verlaufen die Diffusionsvorgänge, welche die Grundlage des Trennprozesses darstellen, bei kleinen geometrischen Abmessungen generell schneller und vollständiger, das heißt dünne Säulen mit geringen Filmdicken an stationärer Phase haben eine höhere Trennleistung.
Zwischen diesen beiden sich widersprechenden Forderungen muss ein Kompromiss gefunden werden. Dieser wiederum ist eng mit dem verwendeten Detektor verknüpft, denn die aus der Säule austretenden Substanzen müssen im Detektor ein elektrisches Signal er-zeugen. Jeder Detektor hat jedoch die allen Messanordnungen prinzipiell anhaftende Eigenschaft, ein Störsignal zu erzeugen, das man als Rauschen bezeichnet. So wie im Radio ein schwacher Rundfunksender im "Rauschen untergeht", verschwindet ein sehr kleiner Peak im Eigenrauschen der Detektoranordnung. Anders gesagt: Empfindliche Detektoren wie zum Beispiel der Flammenionisationsdetektor erlauben die Verwendung von Säulen mit geringer Belastbarkeit und damit hoher Trennleistung, während etwa ein Wärmeleitfähigkeitsdetektor mit seiner um Größenordnungen geringeren Empfindlichkeit die Verwendung von Säulen mit hoher Belastbarkeit verlangt.
Technik der Gaschromatographie
Im folgenden Teil werden die einzelnen Komponenten eines Gaschromatographen und ihre technische Realisierung besprochen. Aus dieser Sicht ergeben sich Hinweise auf deren Handhabung in der Praxis.
"If the column is described as the heart of chromatography, then sample introduction may, with some justification,
be referred to as the Achilles heel"
V. Pretorius und W.Bertsch, HRC & CC, 6/1983, 64
Dieses Zitat illustriert die Tatsache, dass es bis heute keine Patentlösung für die Probenaufgabe in der Gaschromatographie gibt. Das technische Problem, eine kleine Flüssigkeitspor-tion in der Größenordnung 0.1-1 Mikroliter exakt und reproduzierbar in ein System einzuführen, das unter Überdruck steht, ist nicht trivial. Es existieren daher eine Vielzahl von Bauformen und Techniken zur Probenaufgabe, von denen die zwei bekanntesten hier vorgestellt werden sollen.
1. Der splitlose Injektor
Das Prinzip dieser Probenaufgabe ist einfach: Mit einer Mikroliterspritze wird das Septum, eine gummiartige Membran, durchstochen, die Nadel bis zum Anschlag eingeführt und an-schließend der Kolben der Spritze niedergedrückt. Dadurch wird die abgemessene Flüssigkeitsmenge in den Verdampferraum des Injektors gedrückt, dessen Temperatur einige zehn Grad über der Siedetemperatur der Mischung liegen sollte. Der Verdampferraum ist meist eine Glasröhre mit 2-5 Millimetern innerem Durchmesser, die als Liner bezeichnet wird. In der Mitte des Liners befindet sich in der Regel ein Hindernis, das den Flüssigkeitstropfen auf-fangen soll, der bei der Injektion aus der Spritze ausgestoßen wird. An diesem Hindernis soll der Tropfen nun möglichst schnell und vollständig zu einer kleinen Gaswolke verdampft wer-den. Im angelsächsischen Sprachraum wird dieser Vorgang anschaulich mit Flash-Verdampfung bezeichnet. Anschließend wird die Gaswolke vom Trägergasstrom in die Säule fort-getragen, wo die eigentliche Trennung erfolgt.
Diese Art der Probenaufgabe ist mit folgenden prinzip-bedingten Nachteilen behaftet:
• Die Verdampfung des Flüssigkeitstropfens findet nicht momentan statt, sondern benötigt einige Zeit. In dieser Zeit strömt das Trägergas weiter und verbreitert die Gaswolke.
• Das Ausspülen der Gaswolke aus dem Injektor in die Säule ist ein Vorgang, den man mit einer Verdünnungsreihe vergleichen kann. Das Konzentrationsprofil einer injizierten Probe am Ausgang des Injektors zeigt am Ende stets den typischen exponentiellen Verlauf solcher Vorgänge. Dies gilt zwar prinzipiell für alle Probenaufgabesysteme, ist jedoch bei dieser einfachen Konstruktion typbedingt eher zu bemerken.
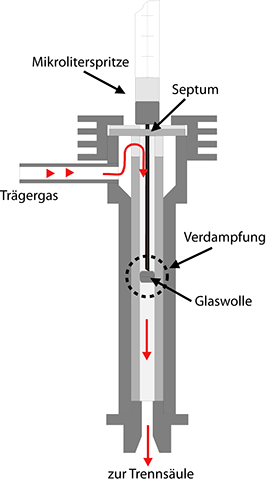
Injektor ohne Split
2. Der Injektor mit Split
Diese Bauform gestattet es, den Trägergasstrom nach der Probenaufgabe zu teilen (to split engl. teilen, spalten). Dazu läßt man einen wesentlich größeren Gasstrom durch den Injektor fließen, als durch die Trennsäule geleitet wird. Abb. 23 soll dies deutlich machen: Zunächst wird die injizierte Stoffportion verdampft. Die Gaswolke wird vom Trägergas weitergetragen, bis sie an den Split gelangt. An dieser Stelle teilt sich der Gasstrom. Der kleinere Anteil strömt in die Säule (meist eine Kapillarsäule), der größere Teil entweicht durch die Leitung und das Splitventil. Das Verhältnis zwischen dem Gasstrom, der durch die Säule geht und demjenigen, der abgeleitet wird, ist das Splitverhältnis (engl. split ratio). Übliche Wert für diese Größe liegen zwischen 1:10 und 1:100. Damit erreicht man zweierlei:
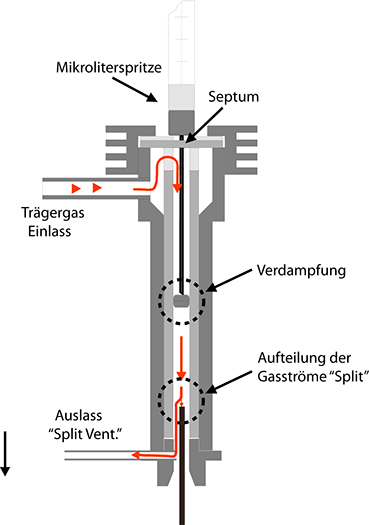
Injektor mit Split
1. Durch die vergleichsweise hohe Geschwindigkeit, mit der die Gaswolke im Injektor an der Split-Stelle vorüberströmt, ist das Konzentrations-Zeit-Profil der zu untersuchenden Stoffe wesentlich schmäler geworden, d.h. die Verschleppung (engl. tailing) des Peakendes durch die Ausspülvorgänge wird stark vermindert.
2. Die Absolutmenge an Analysensubstanz, die in die Säule gelangt, ist um das Splitverhältnis kleiner geworden. Das ist bei Kapillarsäulen eine Notwendigkeit, denn deren Belastbarkeit mit den zu trennenden Stoffen ist um Größenordnungen geringer als bei gepackten Säulen. Ein Rechenbeispiel soll dies noch einmal verdeutlichen: Angenommen durch die Säule fließe ein Trägergasstrom von 10 ml/min, durch den Split-Auslaß ein Gasstrom von 90 ml/min. Wenn der „Gaspfropfen“ nun mit der Geschindigkeit 100 ml/min am Split vorbeiströmt, werden lediglich 10 % der injizierten Stoffmenge in die Säule gelangen.
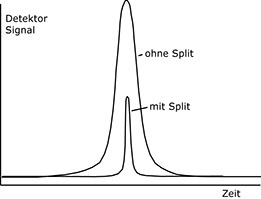
Konzentrationsprofil ohne bzw. mit Split
Wärmeleitfähigkeitsdetektor (WLD)
Der Detektor eines Chromatographen hat die Aufgabe, ein elektrisches Signal abzugeben, wenn eine Komponente des zu analysierenden Stoffes aus der Säule austritt. Es gibt viele verschiedene Arten, diese elektrischen Signale zu gewinnen. Eine davon ist der nun beschriebene Wärmeleitfähigkeitsdetektor.
 Die Fähigkeit von Gasen, Wärme zu leiten, hängt direkt von der Masse der Moleküle ab. Nach der kinetischen Theorie der Gase leiten dabei leichte Teilchen die Wärme besser als schwere. Diese Tatsache wird vom WLD ausgenutzt. Die prinzipielle Anordnung zeigt Abb. 26. Das Trägergas durchströmt von der Trennsäule kommend eine zylindrische Kammer, in der sich ein dünner Draht aus einer Wolframlegierung befindet. Der Draht, Hitzedraht oder Filament genannt, hat einen elektrischen Widerstand R, der jedoch von seiner Temperatur abhängt. Bei Metallen nimmt dieser Widerstand generell mit der Temperatur zu, man spricht von einem positiven Temperaturkoeffizienten (PTC). Wird der Widerstand von einem Strom I durchflossen, so fällt an ihm nach dem Ohmschen Gesetz eine Spannung UDet ab:
Die Fähigkeit von Gasen, Wärme zu leiten, hängt direkt von der Masse der Moleküle ab. Nach der kinetischen Theorie der Gase leiten dabei leichte Teilchen die Wärme besser als schwere. Diese Tatsache wird vom WLD ausgenutzt. Die prinzipielle Anordnung zeigt Abb. 26. Das Trägergas durchströmt von der Trennsäule kommend eine zylindrische Kammer, in der sich ein dünner Draht aus einer Wolframlegierung befindet. Der Draht, Hitzedraht oder Filament genannt, hat einen elektrischen Widerstand R, der jedoch von seiner Temperatur abhängt. Bei Metallen nimmt dieser Widerstand generell mit der Temperatur zu, man spricht von einem positiven Temperaturkoeffizienten (PTC). Wird der Widerstand von einem Strom I durchflossen, so fällt an ihm nach dem Ohmschen Gesetz eine Spannung UDet ab:
UDet = R(T) • IDet
Tritt nun eine Komponente aus der Säule aus, die eine größere Molekülmasse besitzt als das Trägergas, so leitet die Gasmischung die Wärme, die der Hitzedraht liefert, schlechter ab. Seine Temperatur T steigt, damit aber auch sein elektrischer Widerstand. Bei konstantem Strom I nimmt daher UDet zu. Die Anordnung zeigt also Änderungen in der Zusammensetzung des Trägergases als Spannungsänderungen an.
Dieser einfache Detektor hat jedoch den Nachteil, dass jeder andere äußere Einfluss, der die Temperatur des Filaments verändert, ebenfalls eine Änderung der Spannung UDet bewirkt und damit das Nutzsignal überlagert. Beispiele für solche unerwünschten Effekte sind:
- Änderung des Gasstromes
- Variation der Betriebsspannung
- Änderung der Umgebungstemperatur
In der Praxis wird der Detektor daher in dieser Form nicht benutzt. Weitaus günstiger verhält sich die Anordnung in der folgenden Abbildung:
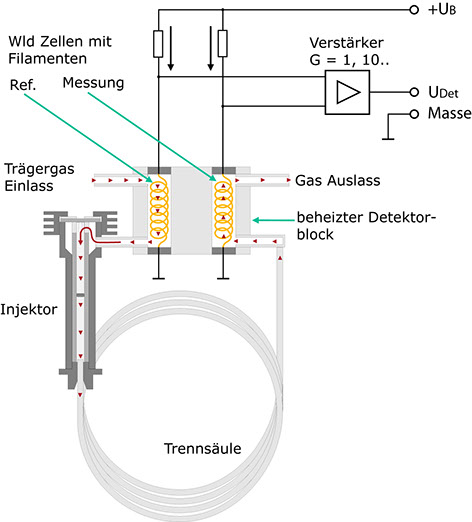
Hier durchströmt das Trägergas vor dem Eintritt in den Injektor eine zweite Messzelle. Beide Messzellen sind zusammen in einem thermisch gut leitenden Metallklotz untergebracht. Auch in dieser Messzelle erzeugt der Strom I einen Spannungsabfall URef am Hitzedraht. Bei Abwesenheit einer Analysensubstanz sollte nun UDet gleich URef sein, wenn die beiden Wolframdrähte identisch sind, und damit auch den gleichen Widerstand haben. Die Brückenspannung UDet-URef müßte Null sein. Bei Austritt einer Komponente aus der Säule gilt dies nicht mehr. Nun ergibt sich zwischen UDet und URef eine Differenz, die in erster Näherung proportional der Konzentration der ausgespülten Komponente ist. Diese Spannung wird verstärkt und in Abhängigkeit von der Zeit aufgezeichnet.
Warum ist dieser Detektor wesentlich unempfindlicher gegenüber Änderungen der oben genannten Größen? Betrachten wir zum Beispiel eine Zunahme der Umgebungstemperatur. Durch die thermische Koppelung der Filamente wirkt sie sich auf beide Filamente zugleich aus. In der Folge nimmt sowohl UDet als auch URef zu, die Differenz UDet-URef bleibt jedoch im Idealfall konstant. Auch eine Änderung des Trägergasstromes hat im Idealfall keinen Einfluß auf die Brückenspannung, weil sie eine Temperaturänderung beider Filamente bewirkt, wodurch zwar UDet und URef verändert werden, die Differenz dieser Größen jedoch annähernd konstant bleibt.
Die wichtigsten Eigenschaften des WLD auf einen Blick:
- Für kleine Konzentrationen an Fremdstoffen ist das Ausgangssignal proportional der Konzentration der Stoffe.
- Die Empfindlichkeit des WLD ist stoffabhängig.
- Die Messzelle des WLD kann nicht beliebig klein gemacht werden. Die Untergrenze für den Durchmesser solcher Zellen liegt bei ca. 2 mm, sein Volumen ist für die Gasströme typischer Kapillarsäulen vergleichsweise groß. In Verbindung mit Kapillarsäulen kann der WLD nur dann erfolgreich eingesetzt werden, wenn diese eine ausreichenden Durchmesser (0,53 mm Innendurchmesser) und eine hohe Filmdicke (z. B. 5 Mikrometer) aufweisen. Der WLD arbeitet zerstörungsfrei, das heißt die eluierten Stoffe werden nicht verändert. Seine vergleichsweise geringe Empfindlichkeit macht ihn wenig geeignet für Spurenanalysen.
- Der WLD sollte nur mit Trägergasen hoher Wärmeleitfähigkeit betrieben werden. Gut geeignet sind Wasserstoff und Helium.
- Beim Betrieb mit einem Temperaturprogramm des Säulenofens muss mit einer Drift der Basislinie gerechnet werden.
- Die Filamente des WLD sind in heißem Zustand anfällig gegen Sauerstoff im Trägergas, der die Wolframdrähte oxidiert und dadurch ihre Funktion beeinträchtigt. Dies kann u.a. geschehen, wenn versehentlich der Trägergasstrom nicht angeschaltet wurde. Daher darf der WLD keinesfalls längere Zeit mit Luft betrieben werden!
Der Flammenionisationsdetektor (FID)
Der Flammenionisationsdetektor ist wesentlich leistungsfähiger als der vorstehend beschriebene Wärmeleitfähigkeitsdetektor.
Bringt man eine Wasserstofflamme zwischen die Platten eines geladenen Kondensators wie in der Abbildung, so fließt in der Anordnung praktisch kein Strom. Enthält der Wasserstoffstrom in einer solchen Anordnung jedoch Substanzen, die C H-Bindungen enthalten, also zum Beispiel Alkane, so nimmt die Stromstärke stark zu.
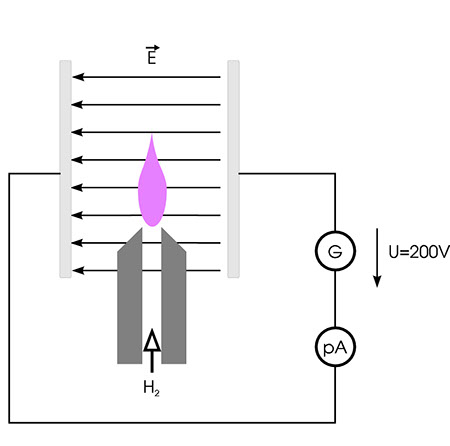
Prinzip des FID
Man führt dies auf die Bildung von CHO+-Ionen und freien Elektronen aus CH·-Radikalen zurück. Allerdings ist hier die Ausbeute an Ionen gering. Im Durchschnitt entsteht nur ein Paar Ladungsträger aus 500 000 Kohlenstoffatomen, trotzdem lässt sich dieser Effekt erfolgreich zur Detektion von Kohlenwasserstoffen und verwandten Verbindungen einsetzen.
Die nachfolgende Abbildung zeigt schematisch die technische Realisierung eines FID. Das Gasgemisch, das analysiert werden soll, tritt dicht unterhalb einer metallischen Düse (1) aus der Kapillarsäule aus. Von dort wird es durch den Wasserstoffstrom in die Flamme geführt. Ihr wird durch separate Kanäle gereinigte Luft zugeführt. Über der Flamme befindet sich eine ringförmige Elektrode (2), die zusammen mit der Düse, der Spannungsquelle (3) und dem Strom-Spannungswandler (4) einen geschlossenen Stromkreis bilden. Dieser Detektor muss beheizt werden, um eine Kondensation der aus der Säule eluierten Stoffe und des entstehenden Wassers zu verhindern.
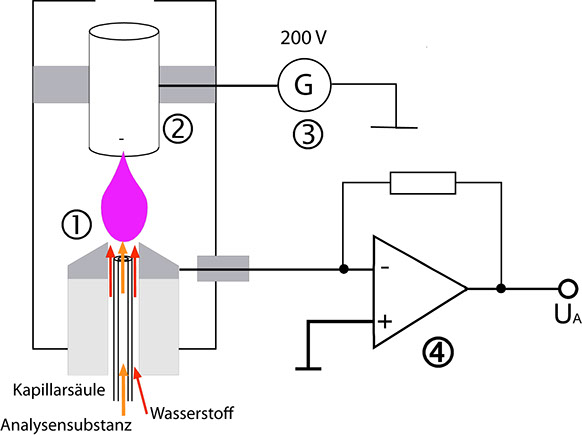
Realisierung eines FID
Die wichtigsten Eigenschaften des FID auf einen Blick:
• Populärster Detektortyp, einfache, robuste Konstruktion
• Spricht nur auf Stoffe an, die C-H-Bindungen enthalten
• Benötigt zum Betrieb Wasserstoff und gereinigte Luft (frei von Kohlenwasserstoffen)
• Muss gezündet werden
• Gute Empfindlichkeit und hohe Messdynamik (Verhältnis von kleinstem zu größtem Nutzsignal)
• Empfindlichkeit abhängig von der Art der detektierten Stoffe
• Relativ unempfindlich gegenüber Änderungen der Temperatur und der Gasströme
Home
Kontakt
Impressum / Datenschutzerklärung